All Features

Rob Moorey
The healthcare technology management (HTM) industry is facing a looming labor shortage and must attract enough skilled professionals to meet future demand. The industry is grappling with an aging workforce, a shrinking traditional talent pool, and increasingly complex medical equipment. Healthcare…

Vick Vaishnavi
The complexity of today’s manufacturing operations and ever-evolving digital disruptions, including the biggest of all, artificial intelligence (AI), require standards that set the pace and keep organizations around the world aligned with what quality looks like.
ISO 9001 has been the…
Quality Digest
(Renishaw: West Dundee, IL) -- The global engineering technologies company Renishaw is introducing its multipurpose RMP400S at the IMTS 2026 and AMB 2026 exhibitions.
The RMP400S seamlessly combines touch-trigger, scanning, and surface-finish verification into a single machine tool probe. This…

Megan Wallin-Kerth
Every industry has blind spots. Sometimes the problem is not a lack of expertise, or even information, but of connection.
While in Austin, Texas, at a recent Octave Live OnTour event, sitting in a large room filled with people from different industries, different methods, different states,…

Megan Wallin-Kerth
Speaking with experts who love their jobs usually makes you aware of two things simultaneously: 1) how much you don’t know, and 2) how much you’d like to know. My conversations with Prashant Darisi and Vick Vaishnavi were no exception. I walked away with an appreciation for the decades of expertise…

Data Physics
Aerospace qualification standards continue to shift as component designs grow more intricate and real-world forces become better understood. Traditional single-axis testing methods don’t capture the complex, multidirectional stresses that modern spacecraft and aircraft encounter. Multi-axis testing…
Sai Ranjith
The U.S. Food and Drug Administration scrutiny of AI and machine learning in medical devices is intensifying. Yet most companies still apply failure mode and effects analysis (FMEA) methods designed for deterministic hardware failures. AI failures are probabilistic, context-dependent, and often…

Chris Chuang
Manufacturers have spent millions investing in safer equipment, smarter automation, and increasingly sophisticated operational technology. Yet employee disengagement, one of the most significant risks to workplace safety and productivity, remains stubbornly difficult to address.
Gallup’s recent…
Mark Stabile, Ridhima Aggarwal
We often think of AI as a technological revolution that will transform industries, disrupt jobs, and change the nature of competitive advantage. But inside organizations, it’s unfolding in a much more complex, less predictable way. In many firms, AI is still more narrative than operational reality…

Mike King, Julie Larsen
Midtier life sciences companies are spending more than ever on quality and regulatory technology, yet many are paying enterprise prices for capability they never use. The right question isn’t whether to invest in a quality management system (QMS) or regulatory information management (RIM) platform…

greenlight.guru
Teams building software as a medical device (SaMD) tend to think of ISO 14971 as the hardware team’s problem. Risk management files, FMEA tables, severity scores: all quality and regulatory territory, while the engineers close Jira tickets. That split is where things go wrong.
ISO 14971 applies to…

NIST
As we celebrate the United States’ 250th anniversary this year, NIST experts have been working to preserve our nation’s history for the next 250-plus years. Among other historic preservation efforts, NIST engineers and scientists created a bespoke time capsule with artifacts from around the country…

Allen Yeung
As a significant portion of the experienced manufacturing workforce approaches retirement, companies face the critical threat of losing undocumented tribal knowledge. Veteran operators possess decades of hard-won, job-specific insights that rarely exist in paper manuals or corporate file systems.…

QT9 Software
For years, many medical device manufacturers approached U.S. Food and Drug Administration (FDA) inspections through a familiar lens: Prepare documents, review subsystem requirements, rehearse likely questions, and demonstrate compliance against a known framework. That approach was shaped by the…

Bryan Balch, Quality Digest
With Hexagon Manufacturing Intelligence’s recent announcement of its inclusion in the NASCAR Competition Partner Program, details on the strategic partnership have emerged. By cementing this tie, the two companies can expand a professional relationship that will enable them to focus on precision,…

Greg Rankin
As pharmaceutical manufacturers push toward lower detection limits, tighter impurity thresholds, and faster development cycles, elemental analysis has become a more critical part of quality and compliance.
Regulatory frameworks such as U.S. Pharmacopeia (USP) <232>, USP <233>, and…

Stephanie Ojeda
In the larger organizations I worked for as a quality leader, supplier auditing was almost always calendar-driven. Sometimes supplier audits happened once a year; in other places, they might happen twice. I’d build the schedule to accommodate that, and the system would just run.
Over time, I…

Aity Ritesh Raj
FDA inspectors don’t just check your records. They bring their own thermometer.
And when their reading doesn’t match six months of logged data from the sensor on your cold room wall, the honest explanation is one nobody wants to say: The monitoring system was compliant; the measurement just wasn’t…

David Isaacson
Quality Digest recently spoke with David Isaacson, the executive director of portfolio marketing at Octave. Besides his work on marketing strategies, he also helps people manage, understand, and better control their operations with software solutions that protect industry assets and people while…

Seb Murray
Industrial robots don’t just shape pay today. Research from Pinar Yildirim, a Wharton professor of economics and marketing, shows they also make workers less likely to move into higher-paying occupations, cutting expected lifetime earnings.
“Workers aren’t necessarily losing their jobs, and it…

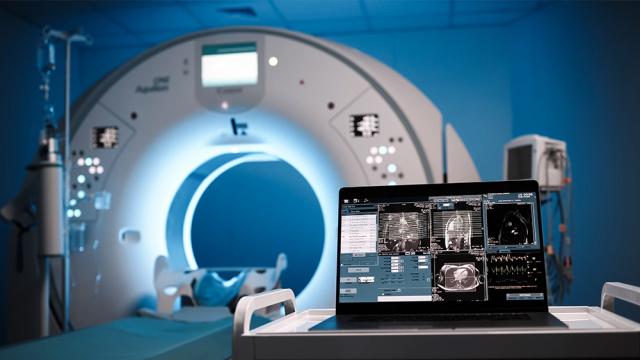
Patrick Gale
Hospitals continue to face increasing financial uncertainty as healthcare reimbursement rates shift and margins tighten.
Medicare reimbursed hospitals at just 83 cents on the dollar in 2024, driving more than $100 billion in underpayments. At the same time, rising labor, supply, and technology…

Arysha Alif Khan
Chemical manufacturing employs roughly 500,000 people in the United States. According to the Bureau of Labor Statistics, workers in that sector noted a total recordable incident rate of 4.2 in 2024, compared to the all-industry average of 3.2. That’s a 32% gap, and it has resisted decades of…

Takeisha Wright
A CAPA investigator opens an AI-enabled quality management system and asks for potential root causes. The system produces several plausible explanations, summarizes similar historical events, and recommends corrective actions. The investigator reviews the suggestions, selects one, and closes the…

Etienne Nichols
The U.S. Food and Drug Administration’s April 2, 2026, warning letter to Purolea Cosmetics Lab is making the rounds, mostly under some version of “FDA cracks down on AI in manufacturing.”
That’s the wrong way to read this. If you build medical devices, treating this issue as an AI story will cause…

MIT News
MIT launched a new initiative—titled Science Is Curiosity on a Mission—to make the case for the long-horizon, curiosity-driven science that has powered generations of American innovation. Through stories of scientists pursuing open-ended questions, the project highlights how fundamental discovery…