All Features

Natalie King
The use of artificial intelligence (AI) is now so widespread that it’s rare to come across a company that isn’t using it in some capacity. While many clients look on the technology favorably, in a recent survey 14% of people still reported that they were unlikely to trust a company that uses AI.…

Andrey Solin
Disclaimer: This isn’t meant to be a car review. This is an article on brand authenticity.
Back in 2021, when Ford was promoting the Mustang Mach-E GT, its high-performance electric vehicle, the company found a way to appeal to potential buyers who somehow missed the sensory appeal of gasoline-…

Annette Franz
Years ago—actually nine years ago, in February 2015—I wrote about “The 7 Deadly Sins of Customer Experience.”
I shared that article on LinkedIn recently and recognized that the sins may need some updating. I don’t disagree with the sins I originally wrote about, but I’m OK with consolidating,…

Mike Figliuolo
It’s all well and good to pontificate about “the customer comes first” or “deliver outstanding service,” but often it’s hard for your team members to wrap their heads around what that really means. You can say these things until your jaw muscles are sore, and post all the customer service rules you…

Gleb Tsipursky
Many employees are unaware that they can leverage the Americans with Disabilities Act (ADA) to request work-from-home (WFH) accommodations based on mental health conditions. This knowledge gap has the potential to reshape the “return to office” (RTO) landscape, creating both opportunities and…

Stephanie Ojeda
Customer complaints are a fact of life in any industry. Even though manufacturers would prefer not to receive complaints, they do come with a silver lining. Managed effectively, they play an important role in improving your products and processes over time—that is, for companies that take a…

Creaform
When it comes to aircraft, poorly documented dents can lead to more significant problems, potentially compromising structural integrity or performance. Dents can trap moisture and lead to corrosion. The stress they generate can initiate fatigue cracks. Their effects on the structure can also affect…

Lawrence Goodman
Here are just a few potential advantages of 3D drug printing—a new system for manufacturing drugs and treatments onsite at pharmacies, healthcare facilities, and other remote locations: chocolate-flavored pills for children who hate taking medicine; several drugs combined into one daily pill for…

Mike Figliuolo
Are you giving your lowest-level employees the power to make crucial customer-relations decisions without supervision? If not, you’re making a huge mistake.
Within a four-hour period on Friday I witnessed both excellent leadership and an abject failure of it. These experiences occurred with the…

Etienne Nichols
If you’re a medtech professional who’s been working with the quality system regulation (QSR) in the United States, then you’re probably familiar with the three terms the U.S. Food and Drug Administration uses for record-keeping requirements: 1) device master record (DMR), a compilation of records…

Bruce Hamilton
As years roll on, I’m noticing more parts of me breaking down: Teeth, eyes, knees, cardiovascular, stomach—the list keeps getting longer, as does the list of docs I see. I’m blessed to be living in an area with the world’s finest medical care and lucky that healthcare innovation (and Medicare) have…

Stephanie Ojeda
Look through even a few FDA warning letters and you’re likely to find violations related to change management.
For instance, a recent warning letter from the U.S. Food and Drug Administration cited a pharmaceutical manager for changing drug components without justification. Another noted a lack of…

AMETEK
Endurance racing is one of the oldest and toughest pastimes in motorsports. It is a true test of performance. Not only is a driver’s stamina on display, but vehicle durability as well.
Based in Indianapolis, Wayne Taylor Racing With Andretti (WTRAndretti) is a world-renowned global motorsports…

Veronica Muzquiz Edwards
Health connects each one of us to one another. No matter where we are in the world, who we are, or what we do, the state of our health is a key determinant in our quality of life. Simply put, it’s our most valuable asset.
Individual health crises can be disastrously grim, and if not addressed…

William A. Levinson
Although quality management has been around in some form or another for thousands of years—a cover of Joseph Juran’s Quality Handbook depicted Egyptians making very precise measurements for the construction of pyramids—this article will show that quality is but one aspect of value, which should be…

Costas Xyloyiannis
To address shrinkflation, by July 1, 2024, stores in France will have to put warning notices in front of all products that have been reduced in size or volume without a corresponding price cut.
“Shrinkflation is a rip-off. We’re putting an end to it,” says France’s economy and finance minister,…

Jim Steventon
Integrated quality, when done correctly, plays a vital and pivotal role in enhancing any business, especially manufacturing operations. But, in fear of sounding like the archetypal head of quality, I say it shouldn’t be seen solely as something you do in manufacturing operations. In fact,…

Daniel Marzullo
When was the last time you asked your clients for genuine feedback on working together, beyond just revisions on projects and deliverables?
Over the years, I’ve found it incredibly beneficial to do quarterly check-ins with every client to get a sense of how things are going.
Not only does this…

NIST
Many consumers across the United States are increasingly aware of the decreasing quantity for many of the products that they regularly purchase and consume. This concept is know as product downsizing or shrinkflation, a term used to describe how a consumer product is sold at the same price, but its…

Gabriel Popkin
They’re called per- and polyfluoroalkyl substances, or PFAS, a group of thousands of compounds that contain a chemical bond between fluorine and carbon. That bond has proved to be one of the most stable and unbreakable known to chemistry—a fact baked into the common nickname “forever chemicals,”…
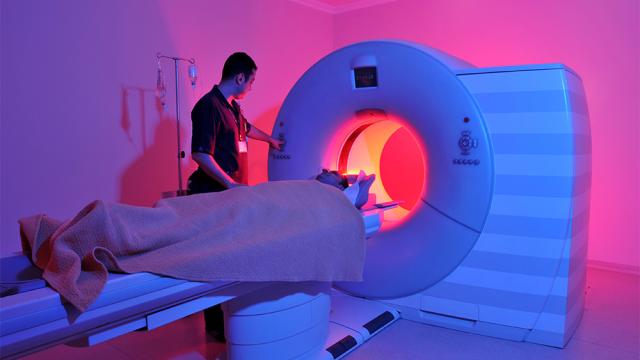
Rob Moorey
Equipment failures in healthcare can have serious consequences, including delays in diagnosis or treatment, scheduling disruptions, and patient safety risks. Health systems should empower clinical engineering teams with technology that helps identify potential failures. This will allow health…

Kate Zabriskie
‘I understand your frustration.” Really? My bank account is overdrawn. I’ve bounced two checks, and it’s because you made a mistake. Unless you’re also having fees charged to your account, I’m not feeling it.
“We apologize for any inconvenience this may have caused.” That’s what I was told after…

Lauren Hinkel
Across the country, hundreds of thousands of drivers deliver packages and parcels to customers and companies each day, with many click-to-door times averaging only a few days. Coordinating a supply chain feat of this magnitude in a predictable and timely way is a longstanding problem of operations…

Mike Figliuolo
I’m fortunate enough to travel to some great places to serve my clients. During those travels, I can’t help but have many customer service interactions from which to draw lessons.
Here, I’ll share how barbecue, airplanes, and coffee can teach you a few things to do (or not do) to create a better…

Roy Arguelles
In today’s marketplace, where products and services proliferate and competition intensifies, businesses are realizing that they must offer more than just commodities to thrive. Enter the experience economy—a paradigm shift where companies are no longer just selling goods or services but crafting…