Social Sharing block
What exactly is a risk-based quality management system (QMS)? This is a timely topic to get into. In 2016, ISO 13485—“Medical devices”—“Quality management systems” was updated, and one of the key concepts presented is the idea of a risk-based QMS.
|
ADVERTISEMENT |
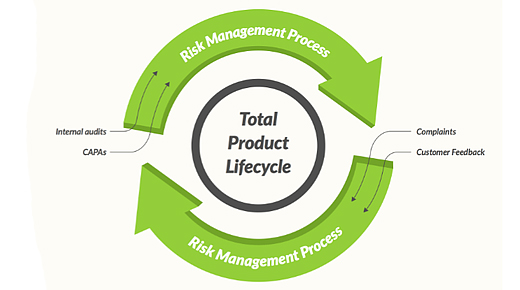
Historically, regulations have almost exclusively looked at risk in terms of either the design and direct product-related elements, or the manufacturing process. Design-focused risk was all about what might happen to the patient, whereas manufacturing-focused risk is the effect of risk elements on our ability to deliver the product. With design risk, you assume the product was manufactured correctly; with manufacturing risk, you assume it was designed correctly.
The new regulations describe the process behind risk management rather than these more traditional product-focused approaches.
Here are some key areas that medical-device developers need to focus on to apply a risk-based QMS.
…
Comments
Risk
Hi Jon,
Thanks for the article. We are in the midst of trying to determine how this requirement affects our QMS. We already have risk incorporated into supplier management, nonconformances, complaints, and CAPA. You started your article with a reference to "ability to deliver the product," but I didn't see that addressed within the article. Also, the standard defines risk as "safety or performance requirements of the medical device or meeting applicable regulatory requirements." Do you have any suggestions related to the other risk items?
Rethinking Risk Management Integration
Thanks Jon,
I was rather ignorant of the "risk" paradigm adopted by ISO 9001, until very recently, which measures "risk" by uncertainty of an outcome, good or bad. Seems more like "measurement uncertainty" than the world we live in.
As we all know Medical Device Regulators we have to meet, and the ISO 13485 standard we now apply are focused on patient risk, first and foremost. Yes, user risks matter too. That is it. I suppose you could link in damage to the environment, as a health risk too.
One of the things I have noted is that the normal approach to identifying, estimating, and mitigating risks, which get captured early in that "risk management file" it often isn't fluid when applied outside the "Design and Development" Process. By that I mean, risk management activities used in the design and development process often don't sync well with other processes, like purchasing, complaint handling and control of nonconforminig product.
I like the article. Your suggestions also point to extending the risk management file out to utilize more features and other documents/forms in other ISO 13485 QMS processes. As this is a brand new standard with so many little changes, its nice to see other articles out their trying to help picture what the future QMS might look like. We need more of this. Thanks!
Risk Based QMS is an illusion
When I first read the 2015 version of ISO's various MMS's I said to myself, what happened to the basics of understanding business management, the customer and interested parties. This new management systems attempts to open the doorway for auditors to step into the board room and start asking questions of the executives and their various planning methodologies. The failure mode is related to the inability of most Quality or Environmental or Safety professionals or auditors to even remotely understand a profit and loss statement or a balance sheet. Most quality or environmental or safety professionals barely understand the financial equation, much less the more finite manipulation of financial activities related to GAAP or financial regulation. I can't wait for the first registrar auditor who attempts to write a non-conformance to the board of directors of an organization. That day will be the beginning of the end of ISO and its various MMS's
What ISO attempted to obtain with its 2015 revision was to force itself into the slot of decision making at the executive level of the business. They will find that to be a territory for which they shall never be welcomed. The strategic motivation for a business shall never be shared with some registrars auditors who inept to sound interpretation of said strategy. THis approach by ISO from their unapproved Guide 83, and the prognostications of Agenda 21, remind me how little these ISO technical committees understand of business overall. QMS was initially a form of supplier control, over time is morphed into a dictated system of business management, written by sources who have little understanding of Strategic, Tactical, and Operational management means. Their lumping of Customers into the basket of interested parties is sufficient evidence that they are actually clueless as to how business actually works. Common sense indicates the fact that without customers, there exist no interested parties
Risk is not a repeatable activity, is one froth with conjecture and supposition. Most quality professionals openly reject the idea of risk for that reason alone. How many risk items related to a PFMEA or FEMA are based upon the consensus of those performing the assessment?
I have discovered after decades of auditing ISO management systems that their purpose is not aligned with the purpose of any given organization. The Technical Committees instead are interested in dictating to the registered organizations what ISO determines they shall and shall not do, including this most recent aspect of forcing the Quality Management System into consideration and control of social media related to the organization (historically a Legal and Marketing position). This is just one example of how ISO technical committees lost focus of their purpose in the construction of this current 2015 revision of its various MSS's
The entire of any QMS can be summed up in comments made by Mr Deming ...and some simple common sense. Fucus upon meeting the needs and Expectations of Customers and Interested Parties, by reducing variation based upon severity of its ipact for an organization to meet said expectations. Within that simple statement any organization can achieve Quality, Meet the Environmental or Safety or Security or Financial regulations, as well as its investors, employees and suppliers.
Tactical subsets of an organization, Quality, Environmental, Safety, Security, Financial etc. should focus upon delivering to the customer a product or service which meets their expectation, while meeting the expectation of Interested Parties usually regulatory in nature. These tactical professionals are not trained to deal with maters of ideological subversion and applied dialectics which have become pervasive via social media and propaganda news outlets.
Marketing/Sales, Executives and Legal council are more suited to addressing social media and other such antithetical war fronts which could effect the public opinion of an organization.
This inclusion of forcing tactical lever managers to respond to ideological subversion and applied dialectics is where the current revision of ISO MSS's have placed a burden upon middle management which is impossible for them to achieve. Assuming that ISO has kicked their way into the board room is not a reasonable expectation, as no registrar is prepared via its audit program to determine if any such actions taken by executive management are either valid or effective. Therefore the current glut of auditors which work for registrars will need be retrained to a level of business understanding not conducive to their historical experience. If we are to take this current iSO revision to heart, gone are the days of a Quality professional becoming a registrar auditor, only those with Board level experience would ever be capable of auditing and with most of those being six figure and stock option entities, I doubt very meny registrars can afford them.
This is why I have great disregard for the current version of ISO's Management System Standards. Their basically, according to their own requirements (19011 etc.) unreasonable and unattainable. Through the political pillar of phiosopha doctrine these prognosticators which are members of the Technical Committees have attempted to wield their sword down upon the Corporations of the world. They shall find the backlash impossible to for them to adequately respond to.
Add new comment