|
|
|
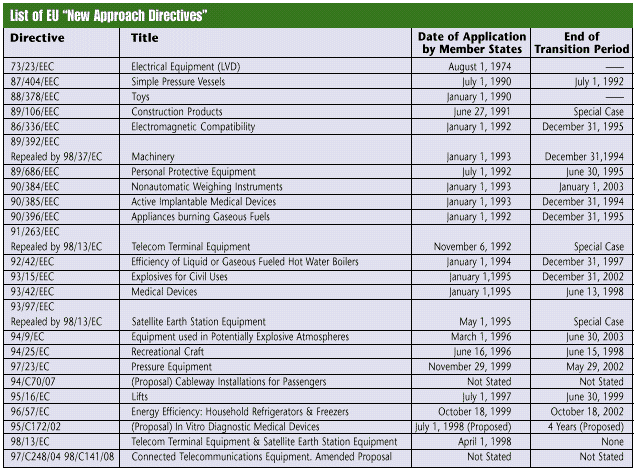
The directives aim to simplify and regulate the movement of goods into and within the EU. In order for the EU to monitor these goods, they require manufacturers to display a CE Marking on applicable products or packaging, and on any accompanying literature. In areas where a directive is in force, it is a crime to place a product on the market without this CE Marking-the manufacturer is legally responsible for ensuring that the product complies with the directive and for placing the CE Marking on the product. The manufacturer also will be held legally responsible for any wrongful placement of the CE Marking on a product. Any directive that applies to the EU and its products is required for any country that sells products there. Thus, as each new directive comes into effect, CE Marking for U.S. goods in that category becomes mandatory. U.S. manufacturers must become aware of the directives and their objectives. The directives are in place to ensure the production of safe devices that are fit for their designated purpose, promote manufacturer responsibility, maintain efficient regulation, benefit the community and control market orientation. Many U.S. manufacturers are finding that they must place the CE Marking on their products to conduct business abroad. The EU is a critical market because it makes up the bulk of the European Economic Area. Established in 1994, the EEA is the largest economic region in the world-consisting of Austria, Belgium, Denmark, Finland, France, Germany, Greece, Iceland, Ireland, Italy, Liechtenstein, Luxembourg, The Netherlands, Norway, Portugal, Spain, Sweden, and the United Kingdom. While not EU members, Iceland, Norway and Liechtenstein are in the European Free Trade Association, which has different rules regarding directives. CE Marking is not a quality standard; it is simply a manufacturer's declaration that a product complies with the essential requirements of the relevant directives. Because CE Marking merely indicates that the product conforms to the minimum legal requirements, it does not ensure that the goods are quality products. It simply allows for the movement of goods and the removal of nonconforming products within the EU. CE Marking is not intended as a marketing tool. In the United States, however, customers often look for the CE Marking on a product to see that it meets these minimum requirements, which other domestic products may lack. The "most popular" directive The Medical Device Directive is currently receiving the most attention. This article therefore uses it as an example to show the steps of CE Marking. The MDD protects patients and operators through the safety, health and quality requirements of medical devices. Its 23 articles explain how it works, with carefully defined steps to guide manufacturers through the conformity procedures. Mandatory in June 1998, the MDD has prompted medical device makers to initiate efforts to affix CE Marking to their products before they lose their eligibility to ship to the EU. The MDD contains four medical device classes-I, IIA, IIB and III. For Class I devices, manufacturers must perform a self-declaration, but no notified body is involved. Class I device manufacturers are on their own, except if the device includes a measurement or sterility function, which requires a notified body to come in to assess the device. For classes IIA, IIB and III, a notified body must be involved. CE Marking basics Obtaining CE Marking can be a long and tedious process, but with the help of a notified body, it need not be overwhelming. Yet sooner or later, companies must undergo the process to continue exporting to the large EU market. For example, the MDD contains five steps: determine classification, meet essential requirements, set up the technical documentation, choose the conformity assessment procedure route and write the declaration of conformity. After completing these steps, manufacturers may affix CE Marking to their medical devices. Before beginning the process, however, manufacturers must first obtain a copy of the directive. (Directives can be found at Web site europa.eu.int/eur-lex/en/search.html . Input the directive number in the search field.) Reading through the document will help them understand what the process entails and what it will mean for selling their product. After examining the 23 articles, the manufacturer should gain a precursory understanding of the process. Beginning the process After understanding the process to follow, the manufacturer begins the classification step. To put the directive into perspective, the MDD is divided into annexes, which are like the chapters of a book and help the manufacturer classify the device. The MDD's 12 annexes are:
In order to determine which stipulations the device must meet and whether it must be tested, the manufacturer starts with Annex IX. This section categorizes the product through a series of criteria. The manufacturer must classify the device or device family based on the classification ruling spelled out in Annex IX. In other words, the manufacturer must read the entire annex and decide which specific rules apply to the product. The next step deals with meeting essential requirements, which Annex I outlines. The manufacturer must read the specifics under Annex I and determine which points apply to the device. Some of these specific classification requirements include device categories such as: chemical, physical and biological; infection and microbial contamination; constructional and environmental; measuring functions; and devices with energy sources. Before moving on, the manufacturer must decide which "harmonized standards" fit with the product. The directives require that whenever possible or appropriate, the manufacturers follow harmonized standards. Harmonized standards can be European norms, British standards or whatnot, but they must be accepted by the EU and published in the Official Journal of the European Communities (also at Web site europa.eu.int/comm/dg03/directs/dg3b/newapproa/eurstd/harmstds ). Manufacturers must identify all harmonized standards that apply to the type of directive they are using-each directive includes a list of those it requires. In other words, the harmonized standards are compiled in a list of norms that have been approved and accepted by all EU members, and the manufacturer is responsible for identifying those standards that apply to them and showing evidence of compliance to those harmonized standards. This information is then compiled in the technical file, the next step in the CE Marking process. A technical file is essentially the evidence that the manufacturer's device meets Annex I requirements. Information in a technical file shows compliance to Annex I Essential Requirements. The manufacturer can determine how to set up its technical file; the only stipulation is that the documentation be available to Competent Authorities (EU members) for review when needed. The notified body simply reviews the technical file, verifies that it has the correct elements and observes that it has a control system.
The notified body evaluates the system against harmonized standards, such as ISO 9000 and EN 46000, to assure that a quality system is in place and all essential requirements are met. The notified body certifies that the product meets requirements by verifying conformity and, if applicable, testing the manufacturer's devices. Again, the designation is based on expertise-a notified body is only designated to conduct a conformity assessment and is restricted to audit and ensure that the product meets the directive's requirements for those areas in which they are certified. Certain notified bodies are not authorized to conduct assessments of certain devices because they do not have the technical expertise on their staff. Also, notified bodies may provide information or helpful tools, but not consulting services. The notified body can guide the manufacturer through the difficulties of the CE Marking process while performing the necessary tasks to complete the process, including performing the assessment to the manufacturer's selected annex route, ensuring the device complies to that annex, notifying the manufacturer that the device has passed successfully and issuing a CE certification. The manufacturer can then begin affixing the CE Marking to the product as well as its packaging and manuals. Further considerations Because the MDD allows for self-declaration for Class I devices, some companies have wrongfully affixed the CE Marking to their device without going through the proper procedures and channels. Either those companies did not understand the directive for self-declaration or they knew that notified bodies were not involved in the process, and without proper guidance, they took the chance that they would not be caught. This only applies to a small group of manufacturers with Class I devices for the MDD, but it nonetheless poses a problem for the EU. Other considerations are language requirements for the countries into which the manufacturer is selling, as well as vigilance systems and complaint handling. Each EU country has its own requirements for handling these processes.Keep in mind that the MDD and others are new directives, with many gray areas. Test cases will determine in which direction they will head, but there are still many conflicting interpretation issues and confusion within the European countries surrounding the directives. Becoming familiar with directive requirements and following all procedures will help alleviate any misunderstandings and keep the process flowing to its conclusion-CE Marking. About the author Korina Ortiz is business development manager for BSI Inc., the North American division of BSI, the world's leading registrar of quality management systems. For more information, please contact BSI Inc. at (800) 862-4977. |
 |
| [QD Online] [Choose CMM] [CMM Guide] [CE Mark] [weibull] |
|
|
|
