by Bevin Hernandez
As readers who work in life science industries know, the U.S. Food and Drug Administration takes quality very seriously. It has regulations for every aspect of business that could affect the quality of pharmaceutical, biotech or medical device products for sale in the United States. In areas such as calibration management, the FDA ensures that systems are in place to control product quality and meet regulations. FDA regulations oversee selecting and implementing many of the computer systems used in calibration and therefore present such implementations with a unique set of challenges.
The upside of such regulation is greater consistency in product quality. The downside includes additional costs and know-how required any time a system is upgraded, even if it's just a minor software release. However, for organizations that make an effort to understand the regulations and the implementation process, these costs can be incorporated into a cost-benefit analysis that can indicate a strong return on investment, particularly given recent developments in calibration and other asset-management systems. The Good Automated Manufacturing Practice Guide 4 (Gamp 4) published by the International Society for Pharmaceutical Engineering ( www .ispe.org ) provides a detailed guide to selecting and implementing regulations and best practices.
As part of the validation process and before an organization launches a new software system, the FDA expects to see documentation of the selection process. One of the first items it looks for is clear documentation of the system's requirements before the selection process. This information is most often found in the user requirements specification (URS), which details what the new system must do and what would be nice if it could do.
Some general requirements for a calibration management system include:
• The ability to add, maintain and retrieve master equipment records
• The ability to add, maintain and retrieve calibration history records
• The ability to track schedules of future calibration due dates
• The ability to print calibration forms
• The ability to compile reports for calibration interval analysis
• Multilevel password protection capability
• Audit trail capability to track database changes
The URS is then matched up with the functional specification (FS) and design specifications, which are expected to come from the software developer. These documents describe the functions of a candidate system and how it was built. When comparing the internal URS to the software vendor's FS, a gap analysis is performed to determine where an internal requirement isn't met, the potential effect of the deficiencies and what possible procedures can be put in place to mediate them.
The organization must also verify that the software vendor has the appropriate methodology in place and is capable of developing high-quality software. The most common ways to achieve this verification include vendor surveys, audits and reviews of vendor-supplied documentation. Software vendors that have experience developing for FDA-regulated companies have established documentation that can greatly speed up this process.
Many of the elements of system configuration and implementation for life science companies are familiar to those outside of life sciences as well. Ideally, much of the groundwork for an implementation is put in place even before the selection process. Often it's wise to perform a baseline assessment--i.e., a discovery of current methods and procedures--so that a clear picture of the effect of the new system can be determined. In a life science context, this assessment is also useful for establishing the URS.
After a final decision is made and the software has been purchased, the organization will want to configure the new system to meet its unique needs and procedures. Depending on the system's productivity-enhancing potential and the software's ability to fit existing procedures, this process will require differing degrees of modifications to those procedures, including developing new ones. For example, when an organization converts from paper-based to electronic signatures, new procedures must be written based on the software's electronic-signature features.
If software is being implemented across multiple sites or distinct groups, it might be necessary to synchronize the system configuration for multiple users. Different calibration information may need to be collected for instruments in the research and development lab vs. the quality control lab or production. Sites may choose different levels of paperless operations, each requiring unique settings. Depending on the products being manufactured, one site might be under higher compliance expectations that will require additional rigor that's unnecessary at other sites. Any number of variations might apply, and the amount of synchronization required will depend on the system's ability to accommodate different configurations as well as how much data sharing and aggregating is desired. The process of deciding where to synchronize across multiple groups and developing the required procedures is often referred to as "user-group blueprinting."
For practical and regulatory reasons, it's important to include data migration in the complete project plan. It's valuable to transfer information from a legacy system over to the new system. In the case of calibration management systems, for example, this means that equipment records are brought over without risk of data entry errors, and the complete calibration history for an instrument can be accessed in one place.
Although many software companies provide services to handle data migration, in a regulated environment a data transfer must be performed in a controlled and documented manner. It's important for FDA-regulated companies to ensure that their data migration plans meet the requirements for a closed and validated system. This includes keeping a strict record of document changes and ensuring that no "back door" exists for data manipulation. If not properly managed and documented, introducing data through an import could be a violation of the closed system.
For life science companies, the data migration plan usually involves three complete transfers of data to the new system. The first is the test transfer, to make sure that all data and details turn up where they're supposed to be. Once all issues have been resolved, another transfer is performed and documented. This transfer is the one used to validate the data migration. Then, just before it's time to transfer to the new system, one last migration is done to capture the very latest information. It's important to select a software system that can accommodate importing data in a controlled manner, and a software vendor that has experience with this sort of migration in a regulated environment.
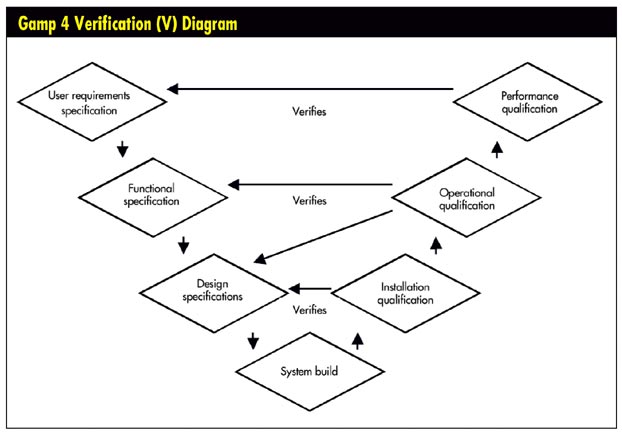
To prove that the system was implemented properly and is working as required and expected, the FDA requires that it be validated. To this end, it looks for documentation that the system, as implemented, meets the requirements specified in the URS as well as those established by the software vendor. As seen in the figure on page 31, the system validation itself is broken into three steps:
• Installation qualification (IQ). This verifies that the software vendor's design specifications were accurate, and the installation was executed according to plan and expectations. IQ documentation includes the procedures for using the software, the hardware that the system runs on, details of the software version and the actual installation, and the library of documentation accompanying the software.
• Operational qualification (OQ). This verifies that the software functions according to the vendor's specifications. The OQ addresses items listed in the FS and elements from the design specifications not covered in the IQ.
• Performance qualification (PQ) . This verifies that the software meets the requirements and expectations established in the URS at the beginning of the selection process. Although the first two steps are generally the same for all companies installing the software, the PQ focuses on how the software is used in a specific context and tests how it performs in that context.
Generating test scripts for validation can be a time-consuming process involving hundreds of different test points. The IQ and OQ scripts, being specific to the software, are often available from the software vendor, especially if it has many life science customers. The PQ scripts, being specific to the company and its operations, naturally must be generated for each company. Some software vendors offer services that will help with this process, combining the vendors' familiarity with the software with experience in what sort of information is required from the site to develop scripts. The collection of scripts and the expected results that would indicate a successful implementation are collectively referred to as the "validation protocol."
Each company must decide the level of documentation that it puts into its validation materials. Generally, there are three types of documentation:
• Pass/fail . Simply indicate for each test if it met the standards established for success.
• Observed results. A description of the behavior is provided.
• Screen captures. An image of the actual screen demonstrates the result of the test.
To determine the degree of rigor required for validating an individual software system, an organization should perform a risk assessment, just as it would for equipment or processes. The FDA's objective is to avoid any intolerable risk to patient safety, and it acknowledges that different organizations and the distinct systems within them pose different risks. Therefore, the FDA has higher expectations for organizations and systems that have a high potential for causing adverse effects than for those that don't have much effect on products that influence human safety. The FDA now encourages a risk-based approach to interpreting regulations and looks for documentation that risk assessments are performed as a justification for the amount of validation executed.
The advantage of choosing a calibration management system designed for life science or other FDA-regulated industries is that the vendor usually will have tools and services available to facilitate the validation. Generating these materials from scratch for software that's been developed internally or from a software vendor that doesn't often sell to life science companies can be time-consuming and require resources that aren't available in many companies.
Given the additional complexities of implementation, particularly those necessary for validation requirements, it's understandable that life science organizations would look for ways to simplify these activities. For some this means simply outsourcing all or parts of the implementation. Many firms can handle the validation. However, few if any independent service providers will be as efficient and effective in performing validations as the software vendor, particularly if that vendor has experience with validations and has helped out with implementation and/or training. Whether an organization performs all tasks internally or outsources them, it's important that a provision is made for project management, which can make a big difference in keeping the project and its costs from spiraling out of control.
Recent software innovations have also reduced the total cost of implementation and validation. For example, many organizations are implementing enterprisewide solutions, i.e., software that allows a single server implementation to be installed at a central location and used at multiple sites. The validation savings of such an implementation can be significant because the server installation and hardware will need to be validated only at the one location instead of at each site. Such solutions, however, are practical only if the software system can control who has access to an individual site's data and allows each site to have its own configuration.
Browser-based applications--where the only software required on client computers is an ordinary Web browser--offer the greatest implementation and validation savings. Until recently browser-based technologies have been slow and awkward, meaning that the validation gains were offset by productivity losses. Software developed on newer browser-based platforms, however, offer the usability and speed of traditional client applications with all the benefits of a "zero-client" implementation, meaning that there's no software to install or download on user workstations.
Another way to simplify implementation and validation is to use a single application for multiple departments, taking advantage of modular add-ons to single-purpose applications (e.g., calibration or maintenance management systems). Until recently, this has meant compromises to best-in-class functionality for one or both departments. To keep best-in-class applications for each department but still receive the benefits of shared information across departments, organizations have commissioned integration between distinct applications. However, this drastically complicates implementing and validating the applications because the two applications, as well as their interactions, must be validated. The latest solution for this challenge are applications designed from inception not as single-purpose applications but as systems that will allow each department to work as it prefers and still retain the collaboration and application demanded by consolidation. Such regulatory asset- management solutions offer implementation and validation benefits without compromising productivity for any department.
Implementation and validation challenges unique to FDA-regulated life science organizations tend to inspire conservative decisions when it comes to implementing new technology, even well-tested and -accepted technologies. However, new regulatory and productivity pressures have motivated these organizations to search out solutions that cut costs and improve compliance. Given an understanding of the cost and resource implications of upgrading, the true short- and long-term return on investment of these technologies can be more accurately estimated and more effective decisions made.
Bevin Hernandez is a project manager for Blue Mountain Quality Resources, a company that has provided asset- management solutions to life science industries for more than 15 years. With each project she balances competing time schedules, budgets, regulatory issues, end-user needs and upper management requirements. More than 90 percent of her projects are at life sciences companies, which keeps her connected with the latest trends and challenges involved with implementing and validating asset-management systems in regulated life science companies. Hernandez has more than seven years of project management experience handling small and large projects in industrial, corporate and educational environments.
|